oxdna2contact.py
This script converts the oxDNA topology and configuration files for a DNA origami nanostructure into a contact map.
Use Non-Relaxed oxDNA Files
The oxDNA configuration file used must be freshly exported from an origami design tool, it must not be relaxed.
Note that oxDNA designs downloaded from nanobase.org have sometimes been relaxed, and thus cannot be reliably converted to a contact map.
Usage:
python3 oxdna2contact.py origami_name
The above command reads the oxDNA files _assets/origami_name.top and _assets/origami_name.dat (or _assets/origami_name.oxdna) and outputs the origami contact map to _assets/origami_name.csv.
After creating the contact map, the script verifies that the contact map passes basic consistency checks. When no sequences exist in the oxDNA files, a warning is generated saying that scaffold-staple base complementary does not exist. However, this is only a warning, and the contact map is still created.
Verify Base Pairings in oxView
The base pairings detected by the algorithm can be visually verified in oxView. This a valuable double-check that the contact map generated is correct, especially if oxdna2contact generated warnings about e.g short staple sections.
To do this visual checking:
Open oxView in a web browser and drag and drop the oxDNA topology and config files into the view area. Using the Google Chrome browser is recommended.
Open the Javascript Console (
Cmd-Option-Jin Chrome on Mac).Locate the file
origami_name.jsin the_assets/directory.Copy the contents of this file, and paste into the Javascript Console.
The paired bases turn pink. Hence, most of the structure will be pink.
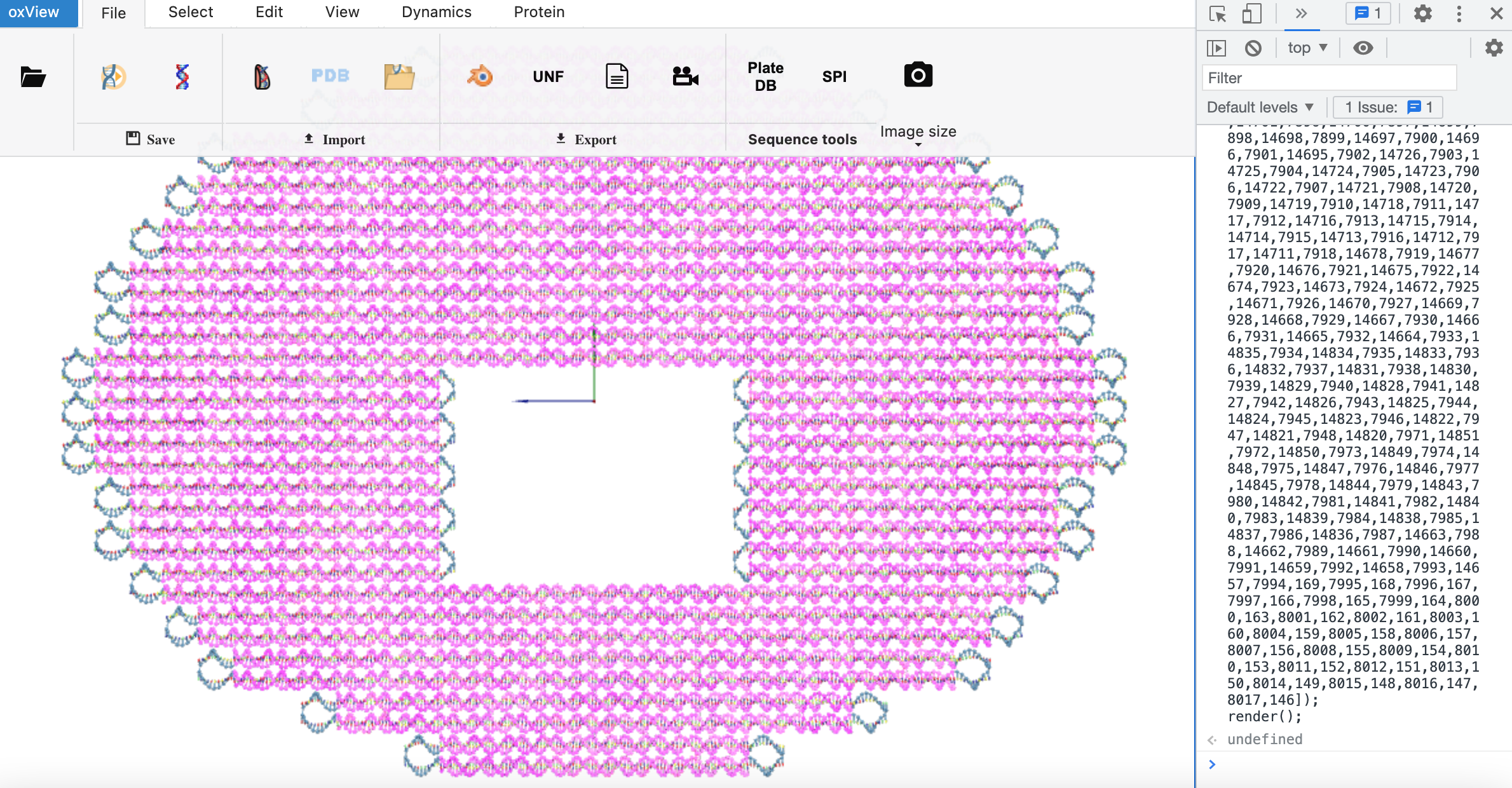
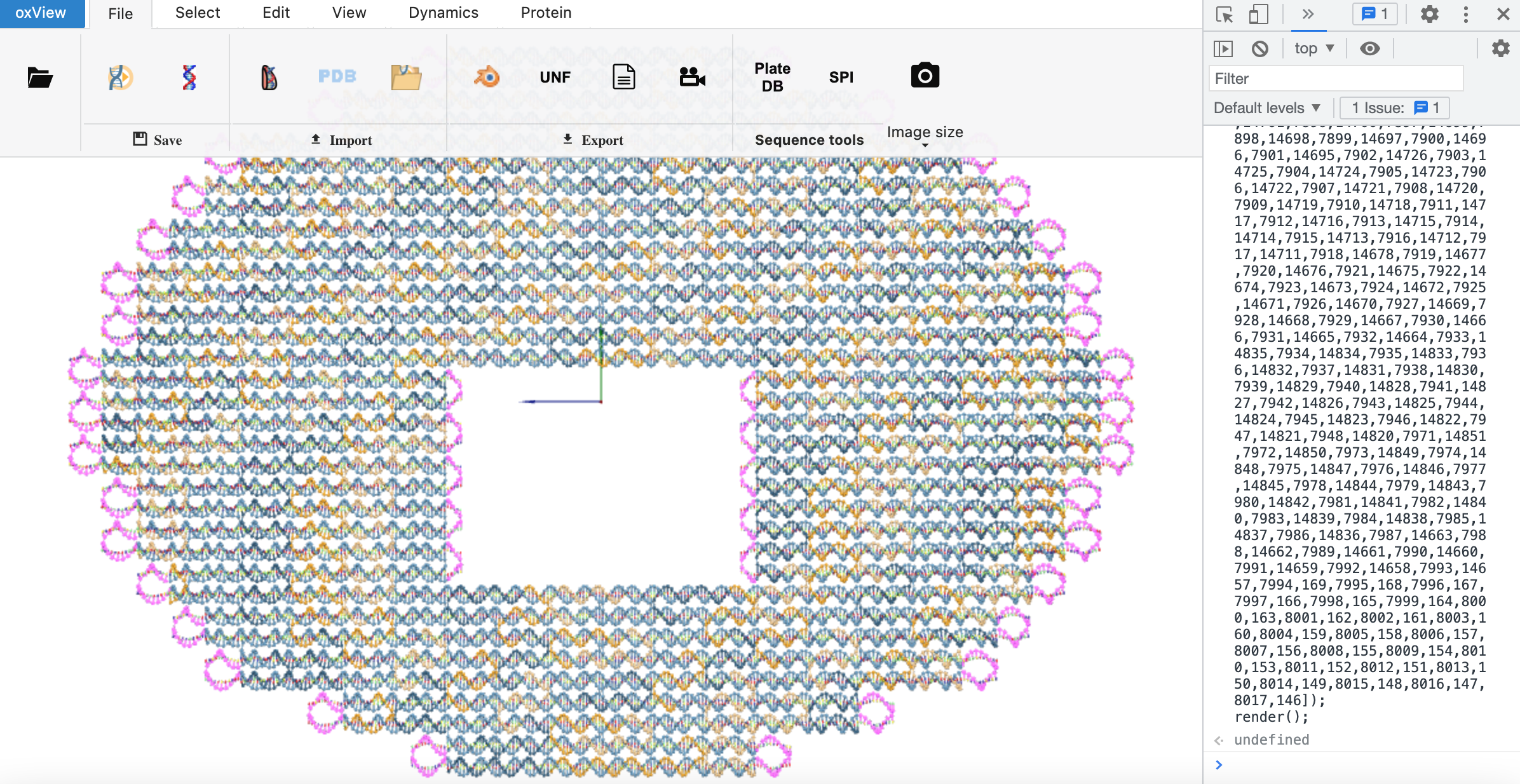
More useful is to see which bases remain unpaired. These should just be single-stranded bases; not bases in helices. To see the unpaired bases, go the the Select tab in oxView and click “invert selection”. Now only the unpaired bases will be highlighted in pink.
Technical Note: Base Pair Detection
The algorithm detects a candidate scaffold-staple base pair in the oxDNA config file when i) the two nucleotides involved are sufficiently close, and ii) both are oriented approximately towards one another.
In more detail, the following three criteria must be satisfied simultaneously:
The distance between the centre of masses (COM) of the nucleotides (r vector) must be equal to or smaller than
EUCLIDEAN_CUTOFFThe vector pointing from the backbone to the scaffold nucleotide COM (b vector) aligns to the vector connecting the scaffold nucleotide COM to the staple nucleotide COM, within tolerance
ANGLE_MAXdegreesThe vector pointing from the backbone to the staple nucleotide COM (b vector) aligns to the vector connecting the staple nucleotide COM to the scaffold nucleotide COM, within tolerance
ANGLE_MAXdegrees
Base pair detection is expressed mathematically as below:


Note
This description of base pairing does not require detailed geometric knowledge of helix particulars like nucleotide shapes, dimensions, and proximity to the DNA backbone. This exact knowledge would be necessary when graphically rendering a strand. But here, it’s sufficient just to know a position indicator (taken as r) and an orientation indicator (taken as b) of each nucleotide, without going deeper.
Technical Note: Fast Base Pairing
For fast base pairing, only staple nucleotides within local a cube region centred around each scaffold nucleotide are tested as being base pair candidates for it. A local cube region encloses a sphere with radius EUCLIDEAN_CUTOFF. Staple nucletides in a cube region are computed by sorting all staple nucleotides in the oxDNA config file in x, y and z coordinates. Then a bisection algorithm is used on the sorted coordinate lists to locate nucleotides in x, y and z ranges of the cube. Finally, a set intersection of the latter three ranges yields the staple nucleotides in a particular cube volume. Once a candidate scaffold-staple pair is found, the next scaffold nucleotide is immediately moved to. If duplicates are found whereby a scaffold nucleotide pairs many staple nucleotides, or vice versa, then pairing fails with an error. Note that the latter situation should not arise if the base pair detection parameters EUCLIDEAN_CUTOFF and ANGLE_MAX are properly set (see oxdna2param.py).
Technical Note: scadnano Loopouts
scadnano allows the special feature of staple or scaffold “loopouts” which are runs of single stranded nucleotides specified as a single crossover (by contrast, caDNAno does not support loopouts). Pictured below is a pathological scadnano structure with a 25nt staple loopout (green) and a 3nt scaffold loopout (blue):

Exporting to oxDNA format, scadnano gives nucleotides in loopouts zero b vectors. These nucleotides thus have no direction, and appear simply as isolated disks in oxView:

As a result, oxdna2contact does not pair any nucleotide with a zero b vector. These nucleotides are simply skipped over.
Comments
The script uses the oxDNA topology file to deduce the scaffold and staple strands, and it uses angles and distances of nucleotides in the non-relaxed oxDNA config file to deduce base pairings between strands (see Technical Note: Base Pair Detection below). Two parameters
EUCLIDEAN_CUTOFFandANGLE_MAXat the top of the script are used to detect staple-scaffold base pairs. Reasonable default values of these parameters have been derived using the oxdna2param.py script with many different origamis.